The FDA has released new guidance on the use of real-world data and real-world evidence in drug development. Why was greater clarification needed, and how can medical affairs drive adoption with this innovative form of evidence generation?
Words by Isabel O’Brien
The relationship between change and risk is often symbiotic: sticking to traditional approaches will likely result in similar outcomes being reached, whereas exploring new routes can lead to transformational results.
But there is a problem when risk could jeopardise an important goal. This is particularly true in the pharmaceutical industry’s drug development processes, where a miscalculation of risk could prevent a new treatment from reaching the bedside of patients in need. This is an issue that has limited the pharmaceutical industry’s use of real-world data and evidence in recent years.
While there has been an increase, a report by Deloitte back in 2020 found that 95% of industry respondents expected RWE to play a significant role in their companies by 2022. This has not happened, with experts suggesting that this is due to a lack of clarity from regulators on their expectations for integrating RWE into R&D cycles.
New FDA frameworks
This could be about to change. Frameworks released by the FDA in December 2021, which cover drugs, biologics and medical devices, seek to provide greater clarity on how pharma companies should be using RWE at all stages of the product lifecycle – from clinical research through to post-approval monitoring.
“The use of computers, mobile devices, wearables and other biosensors to gather and store huge amounts of health-related data has been rapidly accelerating,” says the FDA. “With the issuance of this series of draft guidance, the agency expects that drug sponsors, healthcare providers, patients and the public at large will have a greater understanding of how RWD and RWE can fit into the regulatory process.”
Clarifications on the use of RWE include definitions of appropriate RWD sources, expectations for how data should be analysed and presented so not to favour any particular outcome, and guidelines for general data integrity and criteria to ensure companies have the correct access terms to patient-level data to then use it as evidence at the marketing approval stage.
But that’s not all. The FDA’s guidance also highlights how the development of new and sophisticated analytical capabilities allows for better analysis of RWD and the application of RWE to medical product development and approval.
The series of guidance follows 2016’s ‘21st Century Cures Act’, which placed additional focus on the use of these types of data to support regulatory decision making, and is part of the FDA’s wider, multifaceted Real World Evidence Program. But to what end will this new guidance help to increase the use of RWE?

Expansion of use cases
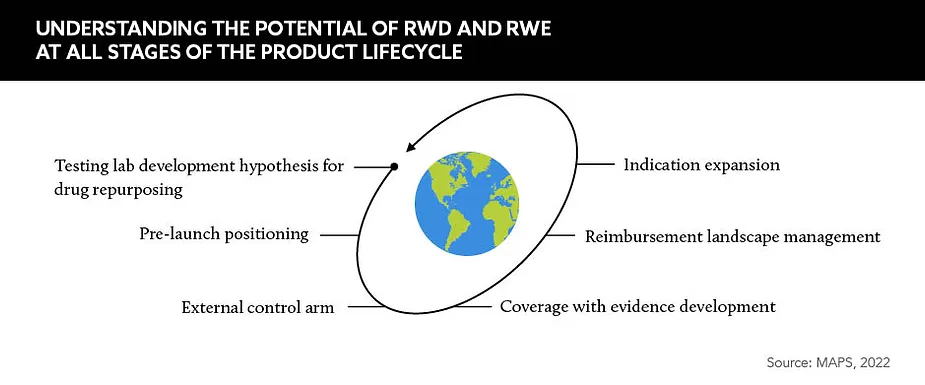
There is potent potential for RWE across the whole product lifecycle, but these frameworks will be particularly useful for companies wishing to harness RWD during the clinical trial phases.
Qin Ye, Principal, Global Real-World Evidence Lead, ZS Associates, highlights an opportunity for overcoming an existing hurdle. “We can leverage RWE to build comparator arms or external controls,” he says. “RWE helps to address a lot of what would be unethical to be addressed through the use of a placebo arm.” When an approval is based on a single-arm interventional trial, RWE can be used as a comparator by companies harnessing historical response rates drawn from chart reviews, expanded access and other practice settings.
RWE helps to address a lot of what would be unethical to be addressed through the use of a placebo arm
In addition, wider use could help to solve existing diversity and recruitment issues that have caused many industry insiders to label the randomised control trial system as broken, ineffective and not serving the needs of patients. The ability to collect RWD outside the traditional clinical trial setting removes barriers such as travel and cost and could ensure that drugs are properly tested within the populations they will eventually serve.
Beyond clinical development, the FDA guidance could enable companies to enhance their use of RWE to understand disease progression. Ye explains that when used effectively, RWE can help in “understanding the real-world effect and how a patient is really responding to the treatment”.
He says RWE can also help to identify disease areas where there is a significant unmet need, enabling companies to not only develop solutions for these populations, but gain insights that could inform effective payer and launch strategies for future indications.
The role of medical affairs
To drive impact across clinical trials, reimbursement and post-approval monitoring, it is critical for companies to understand the possibilities of RWE, but also its strengths and weaknesses at each step of the product lifecycle. Medical affairs must be the key drivers of this understanding, communicating its importance both internally and externally to key stakeholders. This is a capability that is currently lacking across many MA teams.
“Today, in only one of four global pharmaceutical organisations, medical affairs is leading the process of integrated evidence strategy development and planning,” reveals Bora Erdemli, Associate Principal, ZS Associates. Clearly, there is work to be done, but in its position as the “connector of external and internal world as well as development and commercial”, Erdemli believes that MA is perfectly suited to the task of interpreting guidance and building confidence in this form of evidence generation across all functions of a pharma company.
Further progress needed?
There are misconceptions that RWD and RWE could one day operate in direct competition with the traditional randomised control trial, but this is not the case. Rather, the industry and regulatory bodies such as the FDA are waking up to the potential of using the two forms of evidence generation in conjunction. But for regulators in particular, there’s a need to provide clear and detailed instruction to dismantle earlier barriers and deterrents.
The risk is being mitigated, but questions still linger: has this recent guidance gone far enough? And will regulatory applications citing RWE become more comprehensive and widespread in the future? Only time will tell.